Comprehensive Analysis of the Halobacterium Genus through Phylo-pangenomic Approaches
Microbial species delineation has been transformed by the advent of genomics, overcoming the limitations of traditional markers like the 16S rRNA gene. In this study, a comprehensive phylo-pangenomic analysis of the Halobacterium genus was performed using 47 available genomes to assess its genomic diversity and refine its taxonomy. The genus pangenome consists of 13,283 gene clusters, with a core genome of only 827 gene clusters, indicating high genomic diversity. Analysis using Heaps' law yielded a gamma (γ) value of 0.681, confirming that Halobacterium possesses an open pangenome, whose gene pool will expand with the sequencing of new genomes. The phylogenomic analysis, based on 76 single-copy core proteins, coupled with Average Nucleotide Identity (ANI) calculations, allowed for a precise taxonomic re-evaluation. The strains Halobacterium sp. BOL4-2, GSL-19, and CBA1132 were found to exhibit ANI values >99% with Halobacterium salinarum genomes, and their classification within this species is proposed. Conversely, the H. salinarum NRC-34001 genome showed low identity with others of its species 80% and a closer relationship to H. rubrum (83% ANI), suggesting the need for its taxonomic reclassification. These findings underscore the importance of genomic approaches for robust and accurate microbial classification.
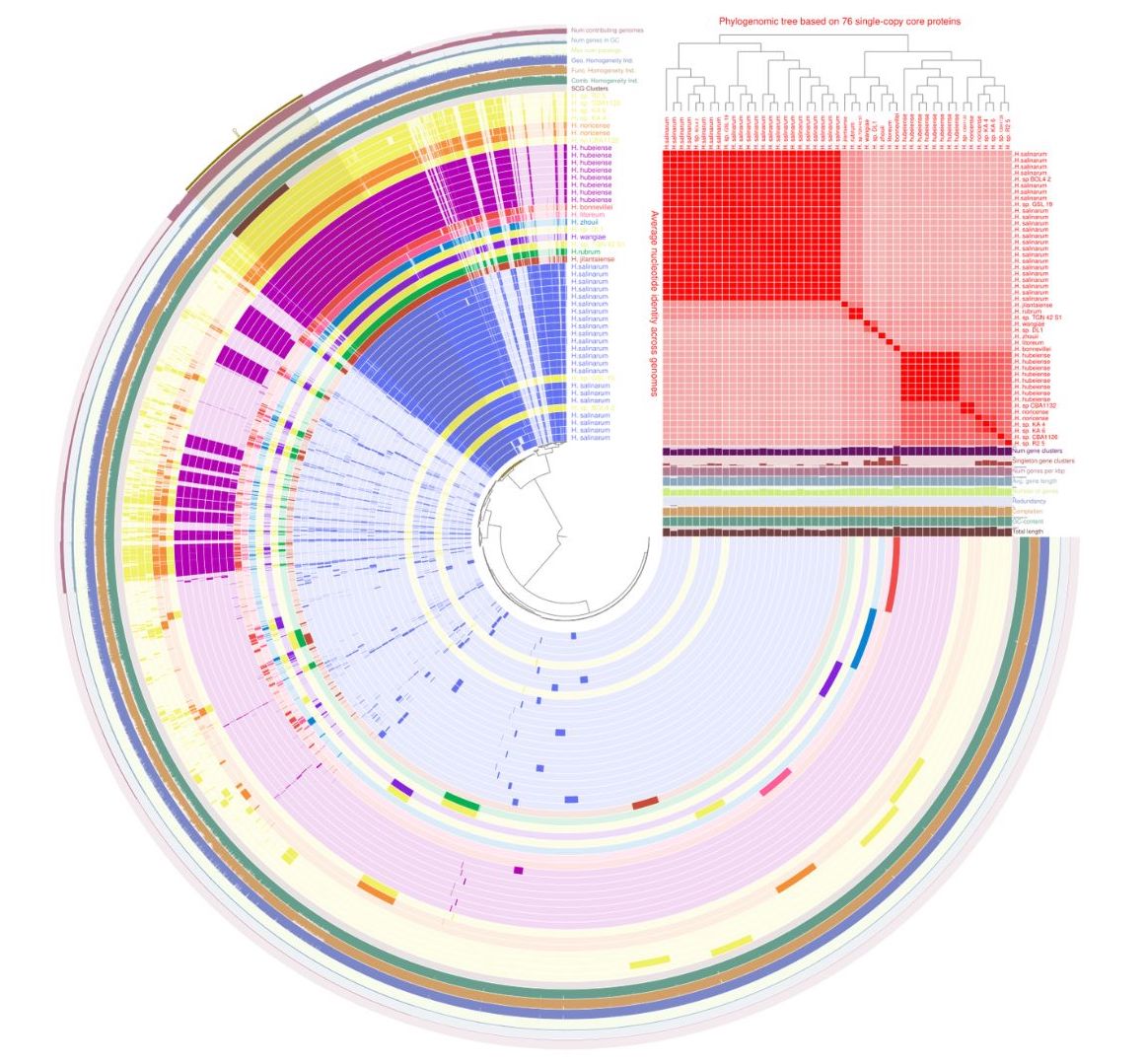
Fig. 1: Pangenome of Halobacterium species (9 species, 47 genomes) generated by anvi’o. The arrangement of genomes is structured according to the presence/absence-based gene cluster tree. Each distinctive color is indicative of a different species. The outer rings encircle supplementary information concerning single-copy gene (SCG) clusters, the count of genes within each cluster, the number of contributing genomes, the maximum paralog count, functional homogeneity, comb homogeneity, and geometric homogeneity. Notably, the outermost red highlights denote the core genes. Additional pertinent details, such as total length, G+C content, completion status, redundancy, gene count, gene count per kbp, singleton gene clusters, and overall gene cluster count, are visually conveyed through the bar charts positioned along the middle-right segment of the figure. Additionally, the top-right corner showcases a heatmap illustrating the percentage identity derived from ANIb analysis with a >95% cutoff and phylogenomic tree based on 76 single-copy core genes for the archaeal domain.
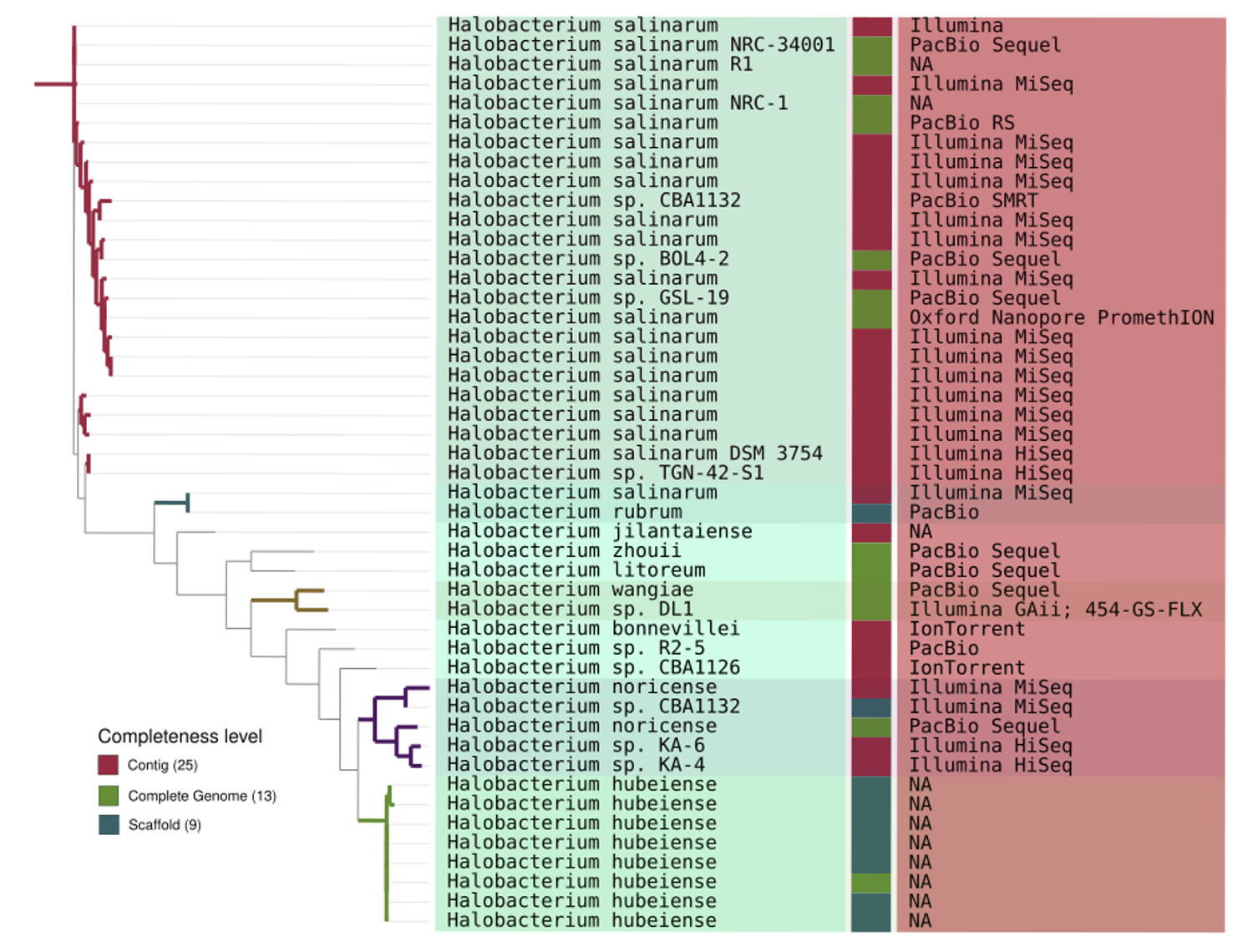
Fig. 2: Phylogenomic tree based on 76 single-copy core proteins. This comprehensive approach encompassed a dataset of 47 strains, each representing distinct genomes from the Halobacterium genus. The figure also incorporates additional data layers, specifically related to completeness level and sequencing platform.